A cooperation between mitochondria and the endoplasmic reticulum is crucial for the mitochondrial health care system called mitophagy
| Journal | Life Sci. Alliance 6(4):e202201640 (2023) |
|---|---|
| Title | The GET pathway serves to activate Atg32-mediated mitophagy by ER targeting of the Ppg1-Far complex |
| Laboratory | Laboratory of Mitochondrial Dynamics〈Assoc. Prof. OKAMOTO Koji〉 |
A process known as "mitophagy" is responsible for the removal of mitochondria, the energy-producing parts of a cell. This occurs if they are defective, or to regulate their numbers. A protein anchored in the mitochondrial surface, called Atg32, promotes this process when it interacts with another protein, Atg11. Modification of Atg32 by "phosphorylation"-the attachment of a phosphate group-stabilizes the interaction between Atg32 and Atg11. The process by which this phosphorylation is regulated was unknown, but now a group from Osaka University has shown that a system known as the GET pathway is required for efficient mitophagy.
Mitophagy requires phosphorylation of Atg32 and a stable interaction between the Atg32 and Atg11 proteins. Mitophagy is suppressed by the action of a protein complex called the Ppg1–Far complex. This acts to reduce Atg32 phosphorylation and interaction with Atg11, and thus suppresses mitophagy.
Proteins located in membranes within the cell must be targeted to their appropriate destinations to maintain the functionality of the different cellular compartments. The GET pathway is responsible for inserting membrane proteins into the endoplasmic reticulum, which is a continuous and dynamic membrane system within the cell. When this pathway is disrupted, proteins can become predominantly inserted into the outer mitochondrial membrane instead.
The team first found that cells lacking the GET pathway showed reduced mitophagy. "This phenotype was rescued when cells lacked both the GET pathway and the Ppg1–Far complex," explains first author Mashun Onishi, "indicating that the reduction in mitophagy that we observed is related to the activity of the Ppg1–Far complex."
"We then went on to show that the GET pathway is responsible for tethering the Ppg1–Far complex at the endoplasmic reticulum membrane," explains senior author Koji Okamoto, "preventing it from interacting with Atg32 to suppress mitophagy, thus allowing Atg32 activation and consequent interaction with Atg11." In the absence of the GET pathway, however, the Ppg1–Far complex is instead targeted to the outer mitochondrial membrane, where it acts to suppress the process of mitophagy.
A protein called Msp1 acts to remove non-mitochondrial proteins from the mitochondrial membrane that have been located there. The team found that disruption of both GET and Msp1 resulted in more severe defects in mitophagy. This suggests that Msp1 might be responsible for removing incorrectly excessively localized Ppg1–Far from the mitochondria and thus maintaining the required levels of mitophagy.
Defects in the process of mitophagy can lead to cell death and have been implicated in aging and Alzheimer's Disease. This work greatly increases our understanding of mitophagy and opens avenues for future research with a significant impact on human health.
Abstract
Mitophagy removes defective or superfluous mitochondria via selective autophagy. In yeast, the pro-mitophagic protein Atg32 localizes to the mitochondrial surface and interacts with the scaffold protein Atg11 to promote degradation of mitochondria. Although Atg32-Atg11 interactions are thought to be stabilized by Atg32 phosphorylation, how this posttranslational modification is regulated remains obscure. Here, we show that cells lacking the guided entry of the tail-anchored protein (GET) pathway exhibit reduced Atg32 phosphorylation and Atg32-Atg11 interactions, which can be rescued by additional loss of the ER-resident Ppg1-Far complex, a multi-subunit phosphatase negatively acting in mitophagy. In GET-deficient cells, Ppg1-Far is predominantly localized to mitochondria. An artificial ER anchoring of Ppg1-Far in GET-deficient cells significantly ameliorates defects in Atg32-Atg11 interactions and mitophagy. Moreover, disruption of GET and Msp1, an AAA-ATPase that extracts non-mitochondrial proteins localized to the mitochondrial surface, elicits synthetic defects in mitophagy. Collectively, we propose that the GET pathway mediates ER targeting of Ppg1-Far, thereby preventing dysregulated suppression of mitophagy activation.
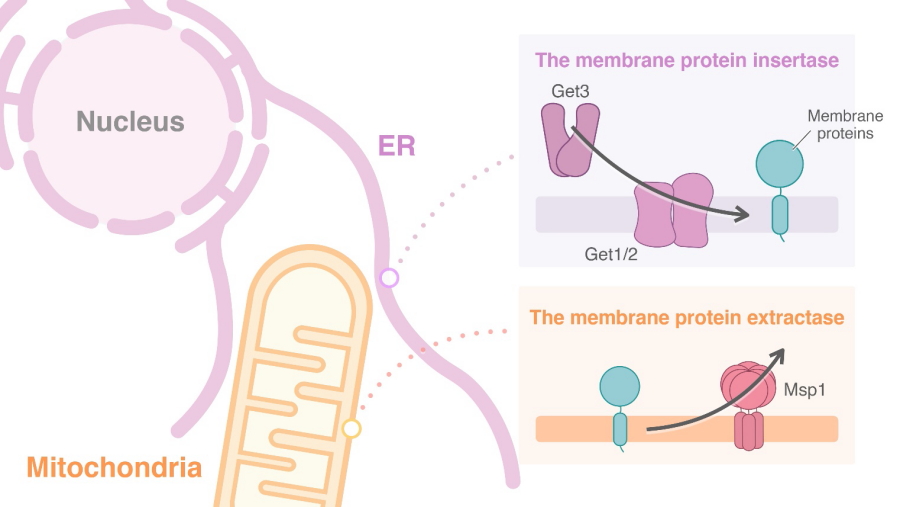
The membrane protein insertase and extractase in budding yeast
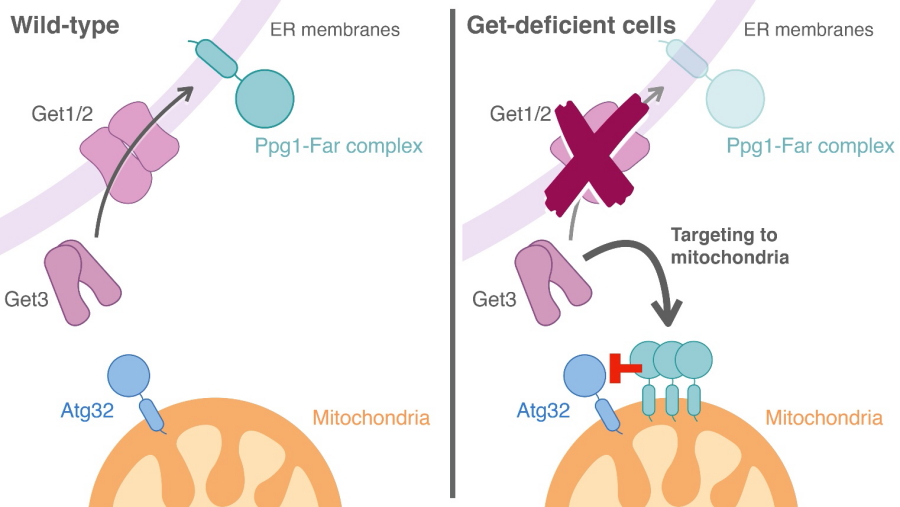
The GET pathway mediates insertion of the Ppg1-Far complex to the ER
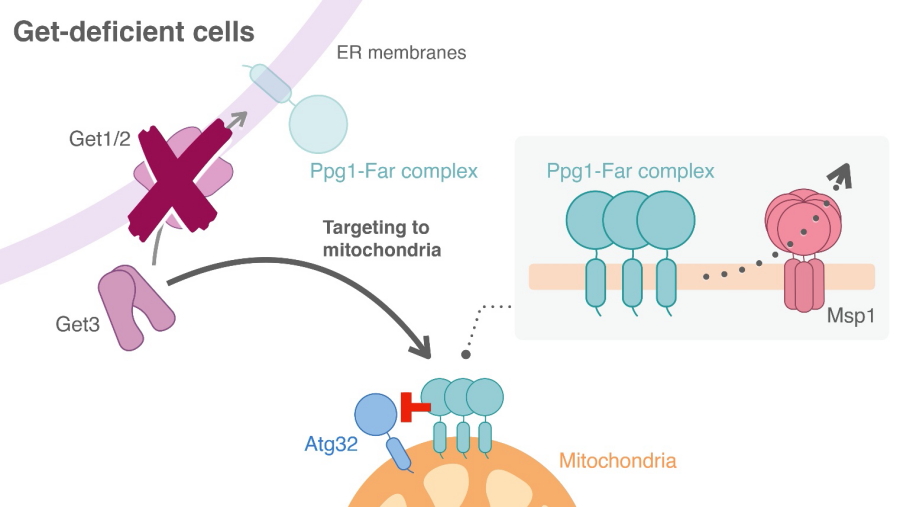
A possible role of Msp1 for removal of the Ppg1-Far complex from mitochondria
| Authors | Mashun Onishi (1), Mitsutaka Kubota (1), Lan Duan (1), Yuan Tian (1), Koji Okamoto (1)
|
|---|---|
| PubMed | 36697253 |