A graduate student visits Prof. Tachibana’s Laboratory
Trying a dry analysis with genome researchers
2021.7.5

With graduate student Ms. MAEDA Hinako, we visited Specially Appointed Assistant Professor MAEDA Ryo and Professor TACHIBANA Makoto in the Laboratory of Epigenome Dynamics (Tachibana Lab). The Tachibana lab studies the role of epigenome alterations in mammalian development and has made important discoveries regarding sex determination in mice. MAEDA Hinako works in the Laboratory for Embryogenesis (Sasaki Lab), where she researches nascent embryos in mice before implantation to study how the body is accurately formed. She is usually immersed in experiments using living things, such as observing cells and studying gene expression. In this edition of our FBS Tour Series, we observed her trying dry analysis--a method that does not use living things and can be only done with computers. Ms. MAEDA thought that it would be difficult, but after experiencing it her initial impression changed.
SA Assist. Prof. MAEDA Ryo (to Ms. MAEDA Hinako): Welcome. Today, we’re not going to look through a microscope but are going to see what we can do with just a computer. Since the development of next generation sequencers, opportunities to analyze huge amounts of genomic data are increasing, so I would hope to think that researchers who conduct experiments on living things (wet analysis) could do dry analysis as well.
Hinako: I've heard about that in lectures. It sounds interesting, though it seems difficult. I’ve asked specialists to do it for me, but I always thought it would be great to be able to do it myself.
Ryo: When you ask a specialist, isn’t it frustrating that it takes a month or two to get satisfactory results? Just like experiments on living things, you cannot get results with only one experiment using dry analysis. It takes many tries to select suitable parameters. When you can do it yourself, you’ll find that it takes less time than asking a specialist and is more fun.

Dr. MAEDA Ryo and Ms. MAEDA Hinako discuss dry analysis
Hinako: How long does it take to create a program to get a complete analysis?
Ryo: Maybe about two or three weeks. This doesn’t require any special sense or talent to get good at. In fact, you don't need to create an analysis program, you just need to want to get results! The key is to be able to maintain your motivation. It’s good to use your own experimental data. What kind of research did you say you are doing, Hinako?
Hinako: My interest is in regeneration and development. Until the second year of my master’s program, I focused on the stage when a fertilized egg progresses from a blastula to a blastocyst and studied the regulatory mechanisms that allow development to correctly proceed. Now, I am researching all the stages before implantation. I would like to understand if dynamic changes that occur after implantation originate in the early embryo, and what the catalyst is for all those changes.
Ryo: So, you would like to know the trigger for drastic changes in gene expression and the mechanisms for development, then. I think I can pull up some familiar data used for a paper we published last year.
Hinako: You mean the paper published in Science, right?

Ms. MAEDA holds the most recent publication: Tachibana Group. "The mouse Sry locus harbors a cryptic exon that is essential for male sex determination" Science, Japanese Scientists in Science 2020, 2021 Issue, March 2021, p49.
Ryo: Yes. It details our discovery of a previously unknown "hidden exon" that encodes the bona fide sex determinant in mice*. I would like to show you how we used dry analysis to make that discovery.
- *The mamalian sex-determining gene, Sry, had been believed to have had only one protein-coding DNA region (exon). However, the Tachibana group discovered a second hidden exon in Sry in mice. They also demonstrated that the second exon encodes a stable sex-determining (male) factor.
Ryo: Using a next-generation sequencer, we obtained the sequence information of all transcripts in Sry-expressing cells and analyzed the gene expression profile. The raw data before analysis looks like this:
Transcription of gene expression (pre-analysis)
Hinako: These looks like DNA sequence alignments, but it seems they would be very complex to analyze.
Ryo: Each sequence (transcript) information consists of four-lines. There are about 40 million sets. We need to clarify where each sequence comes from. It's a lot of painstaking work using spreadsheet software; I wouldn't want to do it. That's one of the reasons why I started learning dry analysis.
Hinako: I see. What kind of coding must you use for the analysis map?
Ryo: We map the sequences obtained from the next-generation sequencer to genome sequences registered in biotechnology databases, such as UCSC and Ensembl. Our code looks like this:

Sample code used in dry analysis
Ryo: We wrote this code to create a file (output) that compares data input (loading data/read sequences) against the reference genome data.
Hinako: The code is simpler than I thought.
Ryo: Yes. This is because researchers in bioinformatics have developed analysis programs that can be easily used by wet researchers. The output looks like this:
The results produced after the program analyzes the gene expression transcripts
Ryo: This is still difficult to work with, so we use a genome browser called IGV (Integrative Genomics Viewer) to visualize the transcription results. It maps where, and to what extent, certain genes are expressed in the genome. Let’s take a look at how Sry was mapped out:
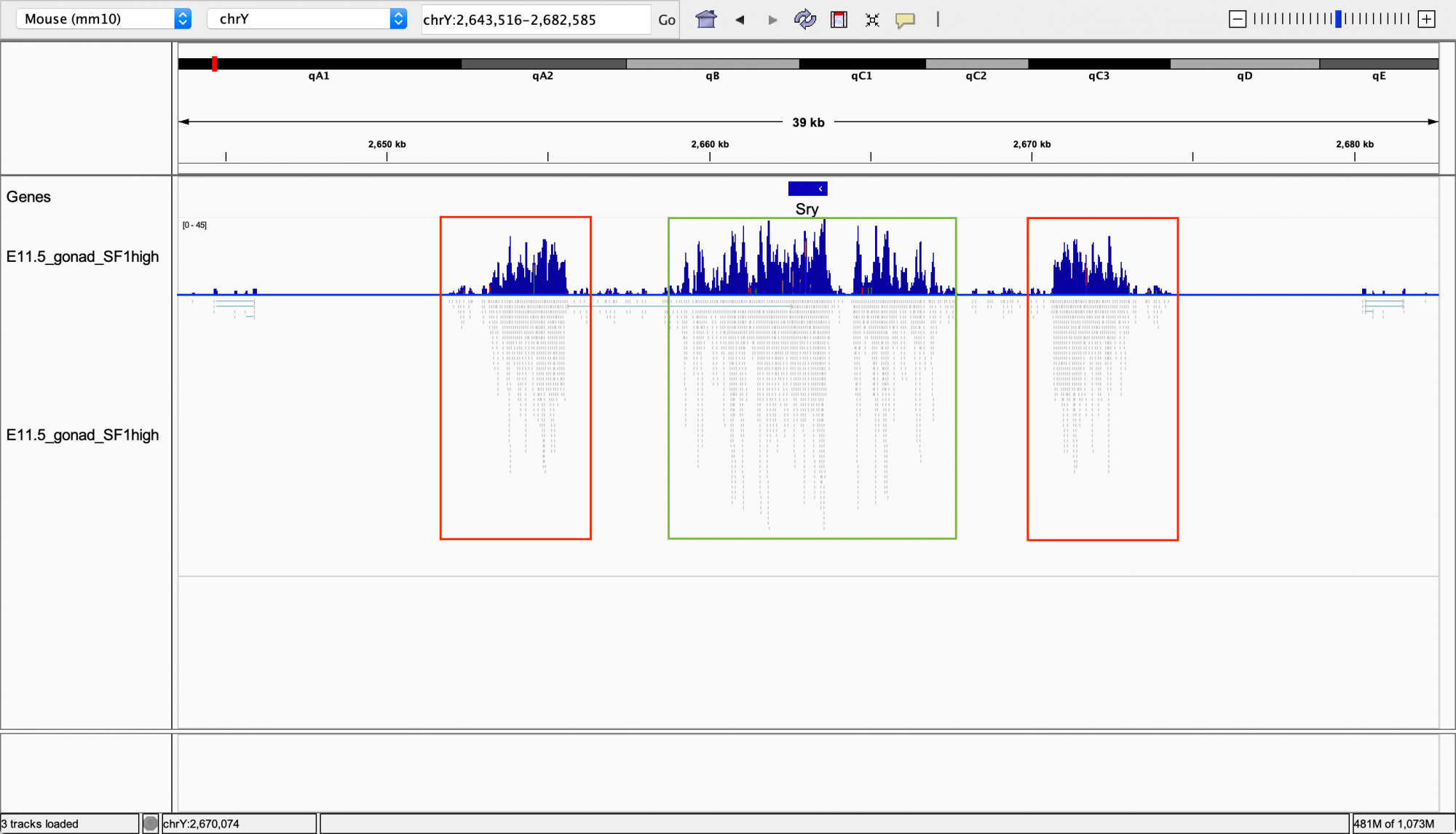
The IGV browser shows its analysis of Sry-expression
Ryo: In the top and bottom rows, the blue line stretching from left to right represents a part of the mouse genome, and the blue histograms show the amount of transcripts.
Hinako: The region marked by the green square is the Sry transcript, isn’t it?
Ryo: Yes, it is. Now, do you see the regions marked in red? A researcher who saw this data said that those regions surrounding Sry were unexpected transcripts*.
- *Sry is unique in that it has a palindrome structure in which the exact same sequence is mirrored on both sides across a known exon. Analyses that eliminate duplicate mappings will not detect mappings to palindromic sequences. After changing the analysis parameters to accept certain duplicate data, the hidden exons were revealed.
Hinako: I get it. Those are the new transcripts you found! These two new red regions show almost the same amount of transcripts as the green one in the middle.
Ryo: Right. Take a look at this figure (below). In addition to RNA sequencing, we performed CAGE and long-read RNA sequencing to determine the starting sites and lengths of transcripts.
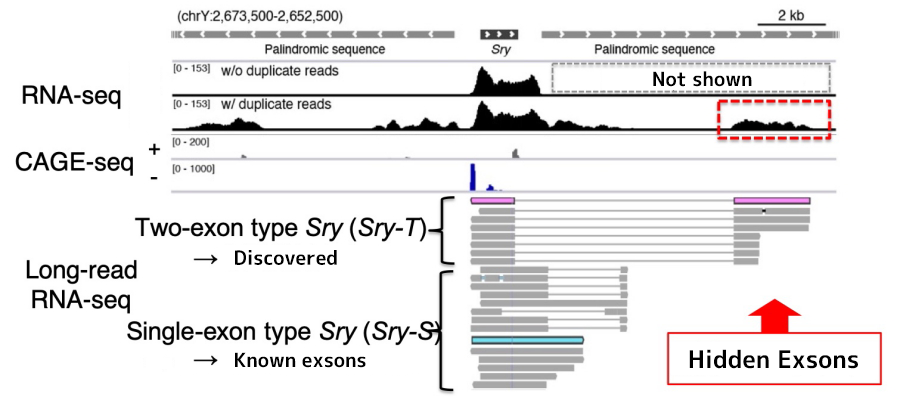
A comprehensive study of gene expression (transcription) in the vicinity of Sry through the extraction of RNA from cells involved in sex determination (fig. above). RNA-seq is an exhaustive study of transcription, CAGE-seq is a study of transcription start sites, and long-read RNA-seq is a study of long transcripts without fragmentations.
Ryo: You can see the location and length of each transcript. In addition to the single-exon type Sry, there is also Sry that is encoded by two separate exons.
Hinako: The portion mapped within the dotted red box are the hidden exons, right? This diagram has always been difficult for me to comprehend when reading research papers, but now I understand it better.
Ryo: Great. So, when we deleted that area, the XY mouse developed as female. Well, it's easy to say, but it's very difficult to delete exons from a mouse!
Hinako: What kind of changes in gene expression did you find in the sex-reversed mice?
Ryo: The following figure shows the comparison of gene expression patterns between males (XY), females (XY), and males in which the hidden exon of Sry was disrupted (SrxKO).
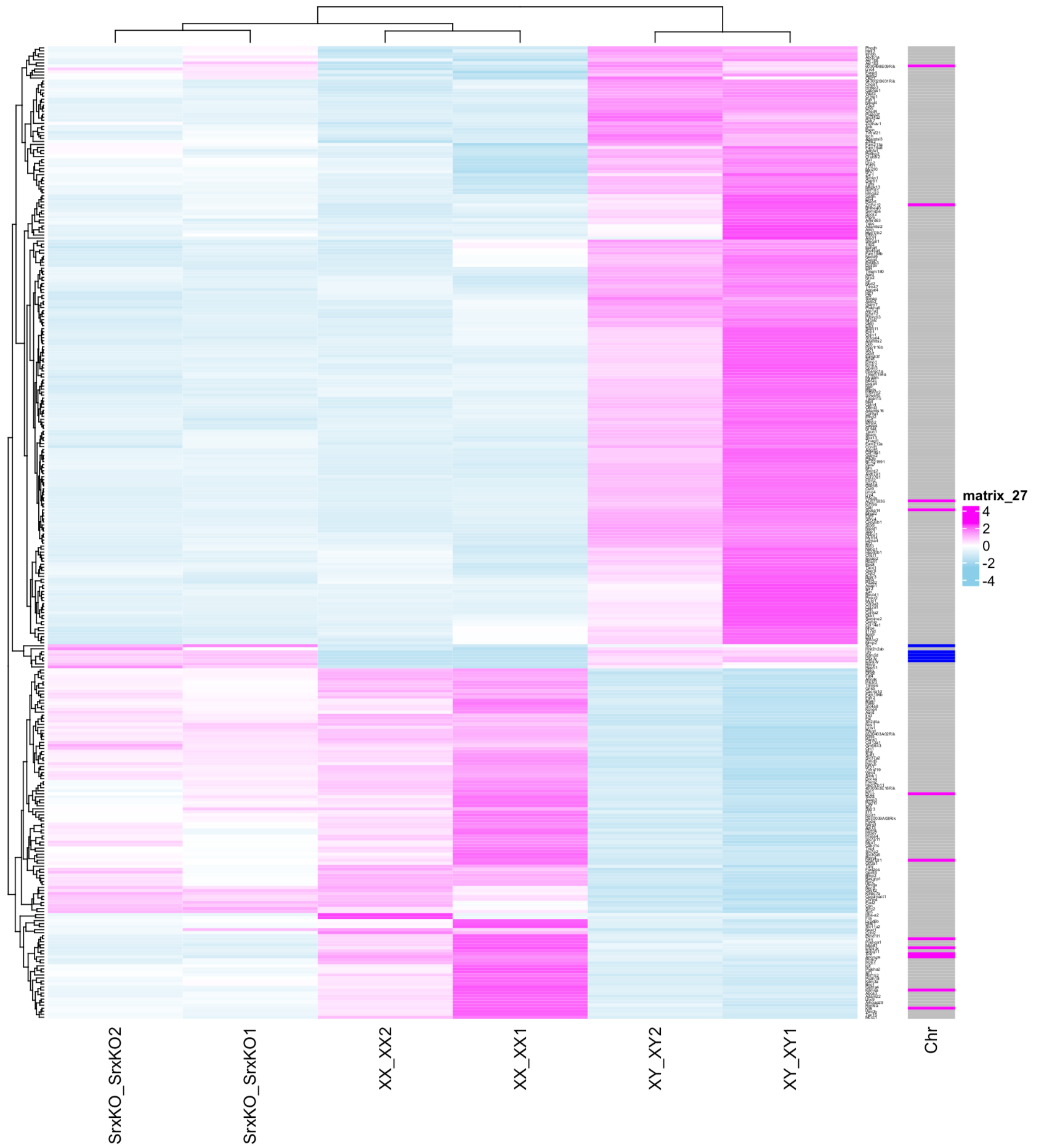
Variations of males (XY), females (XY), and males without the Sry hidden exon (SrxKO)
Ryo: Here, XX, XY, and SrxKO are lined up vertically in pairs. The degree of gene expression is indicated by color. Light blue shows that the gene is not expressed much. Purple shows a gene that is expressed a lot. The two rows on the right are males (XY), and the two middle rows are females (XX). Notice that the patterns in the leftmost two rows (SrxKO…) look just like females even though their genotypes are males. These mice only lack the Sry hidden exon. Though the first exon is expressed, the mice don’t develop into males.
Hinako: That's evidence that the second exon encodes the true sex determinant. It's a beautiful diagram.
Ryo: I think this is the most beautiful figure I have ever analyzed. It didn’t take long to map, quantify, and graph. The mapping process took about two hours to complete three years ago, but now it takes about three minutes.
Hinako: Is that due to the increased performance of computers?
Ryo: Yes, but it could also be due to the improvement of mapping software. To keep up, you have to thoroughly read research papers as the research is constantly progressing. There are also trends in analysis programs and software, and it is sometimes necessary to redo all previous analyses.
Hinako: You’re saying there are research papers that only discuss dry analysis?
Ryo: Yes, sometimes that alone produces results, but more often those papers support research into wet experiments. They do increase the productivity of the lab and also improve my own performance. To be honest, it's very difficult work to do dry analysis while also conducting wet experiments*, but I manage.
- *Ryo’s research is 95% wet. He has taken over the research theme started by the principle investigator of the lab, Prof. TACHIBANA, and is currently studying ES cells.
The first part of the tour complete, we shadow Hinako as she takes the discussion to the principle investigator of the lab, Prof. TACHIBANA Makoto.

Prof. TACHIBANA Makoto, principle investigator of the Tachibana Lab
Hinako: How can I also develop my project enough to submit a paper to Science?
Prof. TACHIBANA: Hmm, that’s a difficult question. I think if you aim to get research results high impact journals are looking for, those results will easily slip past you. Though it may sound simple, what is really important is to do many experiments every day. If you do this, you could come across unexpected results that may make you more curious. Then you just have to follow your curiosity. This is what happened to me and allowed me to develop the paper published in Science in 2013.
Ryo: But I also think it’s because Prof. TACHIBANA won’t let go of something once he’s grabbed on to it.
Prof. TACHIBANA: Right. "If you catch a fish, don’t let it go."
Both Dr. MAEDA and Prof. TACHIBANA shared a laugh at the statement, as they share the same hobby: fishing. They started the "Epigenome Fishing Club" when they were at Tokushima University, and often fished together.
Prof. TACHIBANA: The first author of the "hidden exons" paper, Dr. MIYAWAKI, was also a member of the Epigenome Fishing Club at Tokushima University, although there were only three members. Dr. MAEDA and Dr. MIYAWAKI read the paper published in Science and contacted me to work together. Dr. MAEDA came from Chiba and Dr. MIYAWAKI from Hokkaido. I was very happy that they chose our lab based on its theme and came all the way to Tokushima. Dr. MIYAWAKI transferred to Gifu University, his alma mater, with his "hidden exons" paper as a souvenir last year.
Hinako: It must be great to work with people who have the same tastes in work and hobbies. By the way, do you still do experiments?
Prof. TACHIBANA: I do, yes. Even if someone becomes the principle investigator of a lab, I think if they only write papers, their instincts will dull. I feel sharp when I do experiments. I also have a theme. I worked on DNA cloning the other day and was surprised to see that an easier way to do triple ligation of DNA fragments had been developed, a task which used to be extremely laborious.
Hinako: Do you also do dry analysis?
Prof. TACHIBANA: Ryo is in charge of that. It would be good learn dry analysis when you’re still a young researcher.

Ms. MAEDA interviews the two professors
Hinako: Dr. MAEDA, how long have you been doing dry analysis?
Ryo: I started the programming language, "R," in the second year of my master’s program. Then I learned Perl in I think three years. It will be much easier for you in the future if you learn these languages when you’re still a student. Plus, it’s a good distraction from wet experiments.
Hinako: I’d like to try and learn. What language would be good to start with?
Ryo: There are lots of languages, but if you can master R and Python, just about all languages will be covered. The more people using the language, the better. Python is often used for deep learning AI programs, and R is a common entry language in bioinformatics. It can be run like a spreadsheet, so it is easy to visualize. If you can find a mentor, you can learn faster.
Hinako: Right. I don’t think I’d be as anxious if I had someone I can count on mentoring me.
The conversation regarding analysis diagrams, comparisons of wet and dry research, and even tips concerning Hinako’s personal work continued on, much longer than we could account for here. As both researchers began to relax, we excused ourselves from the lab, but not before Ryo offered one last bit of advice:
Ryo: Dry analysis may solve in an instant what wet researchers are having difficulty with. I hope that in the future there will be much more interaction between wet and dry.
If you are interested in learning more about the activities at the Tachibana Lab, feel free to visit the lab’s websites below or contact Dr. MAEDA and/or Prof. TACHIBANA directly.